Invented by Herman Waldmann, Lisa K Gilliland, Masahide Tone, Mark R Frewin, Louise Walsh, Oxford University Innovation Ltd
Traditionally, monoclonal antibodies (mAbs) have been the primary focus of therapeutic antibody development. These antibodies are derived from a single clone of cells and target a specific antigen. However, the limitations of mAbs, such as their large size, limited tissue penetration, and potential immunogenicity, have led to the exploration of antibody variants.
Antibody variants are engineered versions of mAbs that possess modified properties, such as improved binding affinity, enhanced stability, reduced immunogenicity, and increased tissue penetration. These modifications are achieved through various techniques, including protein engineering, antibody fragmentation, and glycoengineering. The goal is to develop antibodies that are more potent, specific, and suitable for a wider range of therapeutic applications.
One of the key drivers of the market for antibody variants is the increasing prevalence of chronic diseases, such as cancer and autoimmune disorders. These diseases require targeted therapies that can selectively attack diseased cells while sparing healthy cells. Antibody variants offer the potential to achieve this level of specificity and efficacy, making them attractive candidates for drug development.
Another factor contributing to the growth of the market is the rising interest in personalized medicine. With advancements in genomic sequencing and biomarker identification, there is a growing emphasis on developing therapies that are tailored to individual patients. Antibody variants can be designed to target specific mutations or biomarkers associated with a particular disease, allowing for a more personalized approach to treatment.
Furthermore, the advancements in antibody engineering technologies have significantly accelerated the development and production of antibody variants. Techniques such as phage display, yeast display, and hybridoma technology have revolutionized the discovery and optimization of therapeutic antibodies. These technologies enable the screening of large antibody libraries, leading to the identification of high-affinity variants with desired properties.
The market for antibody variants is highly competitive, with numerous pharmaceutical and biotechnology companies actively engaged in research and development. Several antibody variants have already gained regulatory approval and are being used in clinical practice. For example, bispecific antibodies, which can simultaneously bind to two different targets, have shown promising results in the treatment of certain cancers.
In conclusion, the market for antibody variants is expanding rapidly, driven by the increasing demand for personalized medicine and targeted therapies. These engineered versions of antibodies offer improved properties, such as enhanced binding affinity, stability, and tissue penetration. With the advancements in antibody engineering technologies, the development and production of antibody variants have become more efficient and cost-effective. As the understanding of disease biology and biomarkers continues to grow, the market for antibody variants is expected to witness further growth and innovation in the coming years.
The Oxford University Innovation Ltd invention works as follows
This invention is a modified version of an antigen-binding therapeutic antibody, which has a reduced affinity to the antigen as a result or modifications made to the antibody molecules. The antibody can induce immunological tolerance towards the therapeutic antigen. The invention also relates to the method of causing immunological tolerance towards a therapeutic antigen, which involves administering an antibody to the patient that is a modified form of the therapeutic antigen and has a reduced affinity for the cell-surface antigen compared to the therapeutic antibodies.
Background for Antibody variants
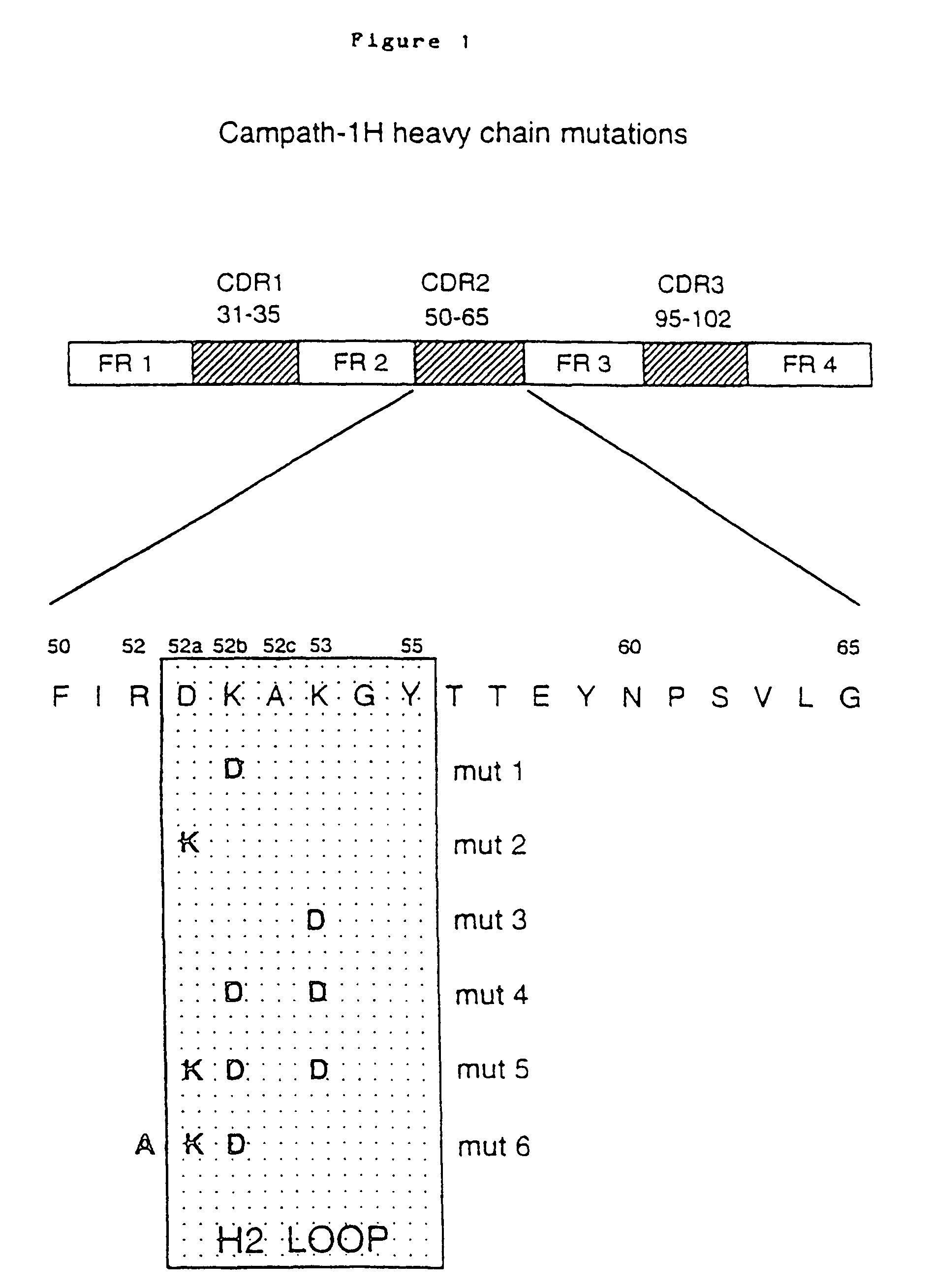
Antibodies that target specific antigens are used to treat various diseases.” Campath-1 monoclonal antibody (mAb) has been used to treat rheumatoid and vasculitis, as well as lymphoma. The target antigen is CD52, also known as CDw52. Xia et. al., 1992) is a GPI anchor glycoprotein found on lymphocytes and monocytes (and in parts of the male reproduction system). It is a peptide of only 12 amino acids with a complex N-linked oligosaccharide attached to Asn3 (Hale et. al., 1990; Xia et. al., 1991). CD52 is an excellent target for antibody-mediated death and can be used in a variety of therapeutic regimens that aim to reduce lymphocytes (e.g. removal of cells from donor bone marrow to prevent graft-versus-host disease, treatment of leukemia and lymphoma, and immuno-suppression).
Several rat anti-human CD52 Campath-1 mAb were generated by fusion of the Y3 rat myeloma line with spleen cells from a rat immunized with human T lymphocytes (Hale et al, 1983). Although the clinical effectiveness of rat Campath-1 mAb has been demonstrated regularly, many patients mounted an anti-antibody (antiglobulin) response against the xenogeneic protein that prevented retreatment with the therapeutic antibody. Antibody therapy is often limited by the antiglobulin response. The anti-idiotypic component (anti-Id; directed against the Ab V regions and in particular the Ab-combining site) inhibits the binding of the Ab to its target while both the anti-Id and the anti-isotypic component (directed against the constant regions) act to accelerate antibody clearance. A major concern is the neutralizing effect of the antiglobulin response. As with antiglobulin responses in general, anti-Id responses interfere with the clinical potency of a therapeutic Ab by forming Ab aggregates that are rapidly cleared from the circulation, reducing the chance for interaction with target antigen. Unfortunately, most antiglobulin sera contain anti-Id antibodies. This has been demonstrated for a number of therapeutic mAb and is especially noted after repeated treatments.
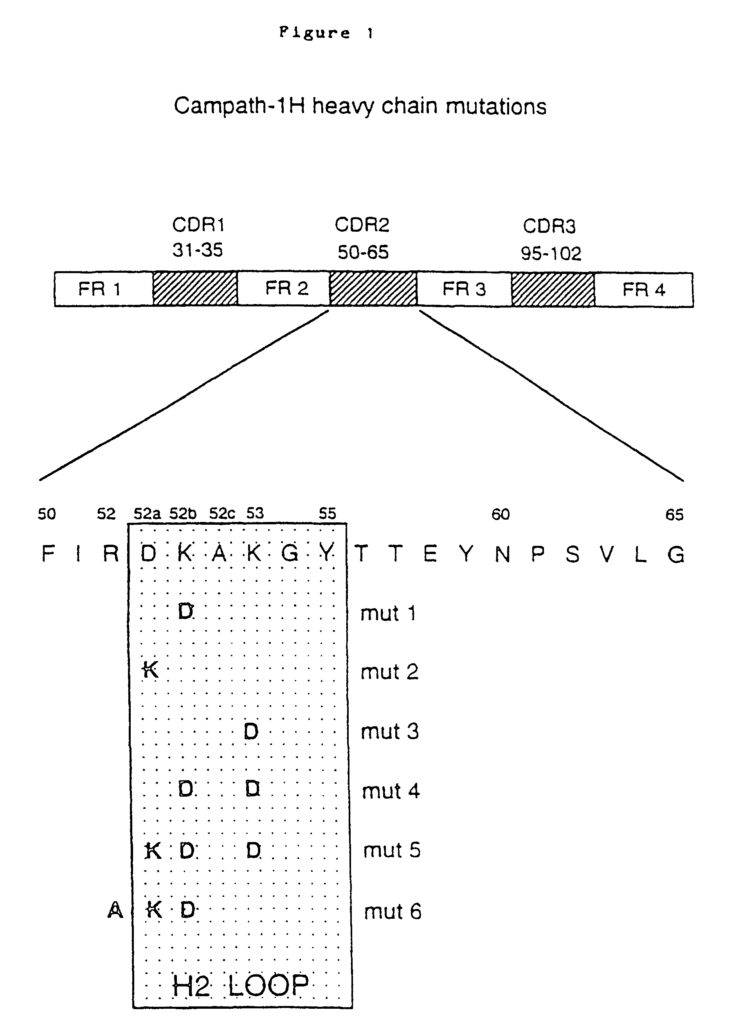
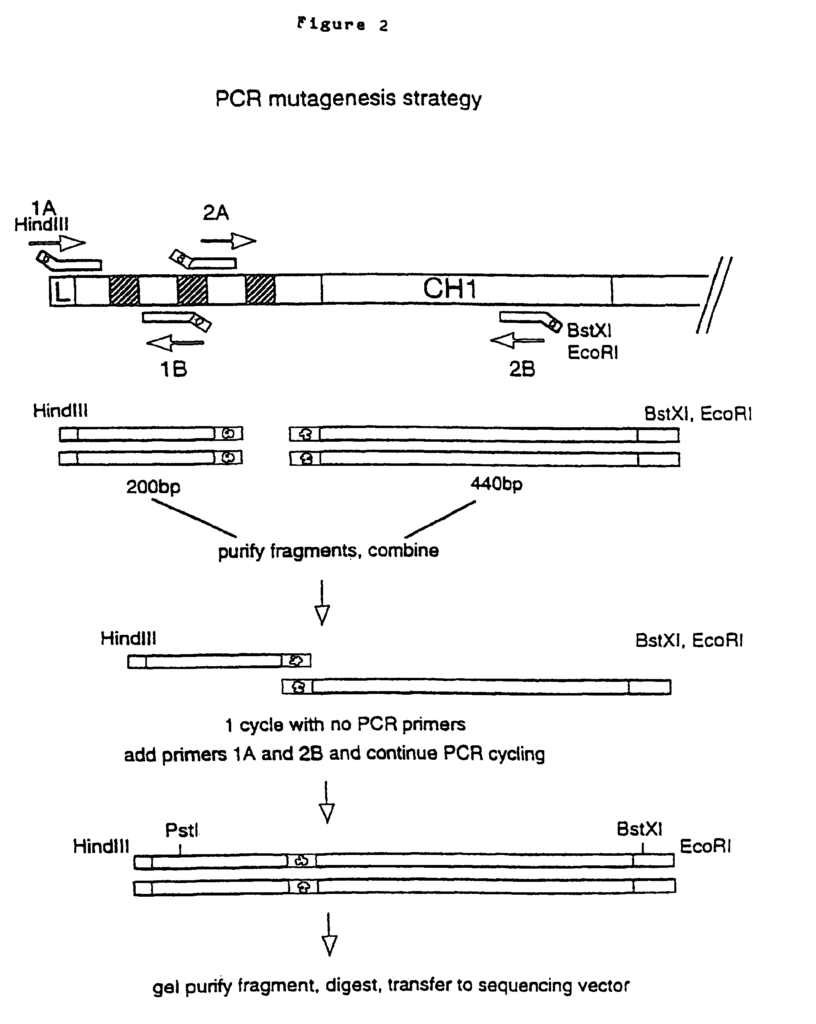
The gene fragments that encode the VL or VH were “humanized” by “CDR grafting”. The rodent hypervariable region was grafted onto the human framework regions by Jones et. al., 1986. Reichmann et. al., 1988. This was achieved by splicing CDR sequences that encoded the rat Campath-1 antibodies onto sequences that encoded human framework backbones provided by crystallographically resolved myeloma protein NEW (for VH) or REI (for VL). Modeling of the humanized V region revealed that the VH framework residue 27 was crucial for maintaining the loop structure in CDR1. The rat residue Phe was substituted for the NEW residue Ser. This resulted back in the restoration of antigen-binding. A second change was suggested (NEW Ser to rat Thr). In functional assays, this substitution did not affect antigen binding. However, the double mutant (Ser27 changed to Phe27, and Ser30 was changed to Thr30), produced the highest amount of protein, and so it was used to create therapeutic humanized Campath-1 Abs, designated Campath-1H by Reichmann et. al., 1988. This change was not deemed to be a risk for the immunogenicity of the antibody, as many human VH frameworks contain threonine in position 30. The humanized VL, VH and constant regions were genetically fused to the human light chain or heavy chain. The humanized Campath-1 antigen consists of human residues in all positions, except for those that encode the three CDRs (light chain) and the three CDRs (heavy chain), and the residues Phe27, and Thr30, found in the VH heavy chain.
In clinical trials, it was discovered that the humanized version of the Campath-1H antibody (rat IgG2b Campath-1) is much less immunogenic. Humanization decreases the immunogenicity in rodent mAb. However, both the allotype and idiotype of humanized mAb may still be targets for humoral reactions. Some allotype-matched Campath-1H recipients have shown idiotype sensitization (Isaacs, 1992; Lockwood, 1993). The presence of anti-id antibodies in patients’ sera revealed these responses. One patient produced high-titre anti Id which cross-reacted with the entire panel CD52 mAb. In EP 0328404, humanized Campath-1 antibody is described. The teachings are fully incorporated into this application by reference.
A strategy to further reduce immunogenicity of Campath-1H could be to regraft the six CDR loops on well-characterized human Germline Framework Regions. Most humanized V regions have so far used rearranged V genes as acceptor sequence. Campath-1H was an example, as myeloma protein framework sequences were used to create acceptor sequences both for VH and VL. Rearranged V genes often contain somatic mutants acquired through affinity maturation. They will be specific to the person from whom the rearranged gene was derived, and may therefore be perceived as foreign by another individual. Regrafting could introduce new idiotypic epitopes – formed by junctional regions that encompass CDR residues as well as new framework residues. Humanization may not be enough to solve the anti-Id problem, as the human population has outbred itself and is unlikely that every patient will tolerate a humanized mAb. There are many different alleles that carry allotypic marks to which natural antiglobulin reactions can be demonstrated, even in constant regions of antibodies. “The problem is more complicated for V-regions, which have a greater degree of variability in both allotypes and haptotypes compared to constant regions.
Another way to achieve this is by inducing tolerance to the foreign peptides that are contained within the Campath-1HV-region. We know that antiglobulin is a B cell response, which is dependent on CD4+ T cells. Isaacs and Waldmann (1996) demonstrated that mice devoid of CD4+ T-cells could not respond to a mAb binding to foreign cells (rat anti-mouse CD8-mAb). Depletion of CD4+ Tells was achieved by adult thymectomy in conjunction with the administration of depleting CD4 antibodies. The response of these mice to SRBC or mAb was assessed. CD4+-deficient mice did not produce either an antiglobulin or anti-SRBC reaction, demonstrating the classical dependence of the anti Ig response on CD4+-T-cells. To generate T-cell assistance and get the correct T-cell reaction, the administered Ab must be treated as a protein-antigen and presented by an antigen-presenting cell, most likely in the context with an MHC class-II molecule. Two main strategies are available to reduce the immunogenicity a humanized V region. 1) We can “silence” (1) We can?silence?
?Silencing? “Antibody Molecules:
Theoretically, we could silence the antibody so that the immune response will not recognize foreign determinants. We could do this if we scanned the VL or VH amino acids for motifs that would bind to MHC Class II molecules. By site-directed mutation, we could identify the key residues in a class II peptide which were not responsible for antibody affinity or specificity. T-helper peptides do not occur randomly, and each protein only has a small number of peptides that can bind to MHC class 2 molecules and to T cell antigen receptors. This is not currently possible because the class II-binding proteins are not sufficiently characterized for them to be identified through scanning of protein sequences. The heterogeneous nature class II peptides is partly responsible for this. In comparison with processed peptides from MHC Class I molecules, naturally processed peptides from MHC Class II molecules tend to be larger and more variable in size. They also have ragged ends on both the C- and N termini. MHC class II associated peptides vary in length from 8 to 24 amino acids. (Rudensky, Janeway and Hunt, 1993; Hunt, et. al., 1991). The sequence motif of class I peptides has specific anchor residues at certain positions that allow their side chains fit into the binding pockets in the peptide binding groove. This groove is also closed on both ends. Class II peptides, on the other hand, are bound in a conformation that extends from both ends of a?open ended? The antigen binding groove is ‘open-ended’ If we were able to predict the class II peptide binding motifs, other strategies could be used to?silence? You could, for example, insert a protease-cleavage site into any class II epitope in order to increase the likelihood of degradation of the peptide before it is presented within the context of Class II. Inserting motifs such as Gly/Ala repeats into the V-region may also inhibit degradation into peptides which could be associated with class II molecules. In one system it was demonstrated that EBNA1 repeats produced a cis acting inhibitory signal which interfered with antigen presentation during MHC class I restricted presentation, thereby inhibiting CTL recognition (Levitskaya et. al., 1995). Both approaches have some potential in the future. However, they rely on the prediction of MHC class 2 peptides based on protein sequences of humanized VL/VH regions. This is limited by a lack of knowledge about consensus motifs of class II epitopes.
In the absence of sufficient knowledge about class II peptides, we have focused our attention on induction of toleration for therapeutic antibodies. Benjamin et al. and Cobbold et al. described a surprising property of cell-binding mAb in 1986: whereas tolerance could be induced to the Fc (anti-isotype toleration), the idiotype remained immunogenic under similar conditions. It was relatively simple to induce tolerance for non-cell-binding mAb, but cell-binding antibodies were very immunogenic.
Isaacs & Waldmann (1994), in a preliminary investigation, used a non cell-binding’mixed molecules? Tolerance to the wild type form can be induced by derivatives of an Ab that binds to cells. The anti-CD8 mouse model was used to produce the cell-binding antibody. The non-cell binding derivatives were created by pairing relevant H- and L-chains to an irrelevant L- or H-chain. By limiting dilution-cloning the hybridoma expressing the anti-CD8 L- and H-chains, as well as a myeloma-light chain (from the partner Y3), we obtained the relevant H-chain. The hybridoma was cloned to express the anti-CD8 specific H-chain, but not the anti-CD8 L chain. This method also produced a clone that expressed only the relevant L chain. This clone, which expressed an irrelevant specificity of anti-human CD3, was fused with a hybridoma. A variant was selected expressing the anti-CD8 H-chain and the anti-CD3 L-chain. Since proteins are converted into peptides before being presented to T-cells they would be “seen” by the T-cells. T-cells will recognize helper peptides from antigen-specific H and L chains, regardless of the partner chain. In this case, however, there was no benefit in tolerance induction when using non-cell binding mixed molecule derivatives from a therapeutic anti-mab in vivo as compared to matched isotype controls, suggesting that most (or even all) helper epitopes are located in the constant region.
In practice, it would not be possible to use these “mixed molecules” It would be impossible to use antigen-specific immunoglobulin chains with irrelevant immunoglobulin chain for human therapy, because the irrelevant H and L-chains carry helper epitopes. This would make it difficult to achieve tolerance towards the relevant H and L-chains. One would not expect to tolerate B-cells that’see’. “Idiotypic determinants are formed by the combination between the H- and L chains of the antibody.
Campath-1, a cell-binding therapeutic mAb, would benefit from a tolerogen that is effective with it. This could be a non-cell binding form of the therapeutic antibody.” It is the same for other therapeutic antibody variants that have cell-binding characteristics.
The Invention
The invention provides an antibody that is a modified form of a therapeutic antigen with affinity for a surface antigen of the cell, with reduced affinity compared to the therapeutic antigen as a result or modifications to antibody molecules, and wherein the antibodies is capable of causing immunological tolerance to therapeutic antigens.
Preferably the affinity of antibody according to invention for antigen is reduced by 50% or less compared to the affinity for antigen of therapeutic antibody. The affinity is preferably reduced to 10%, or 1%, of that of the therapeutic antigen. The affinity must be sufficiently reduced for the antibody to function as a tolerogen in relation to the therapeutic antigen. The term “non-cell binding variant” is used. The term “non-cell-binding variant” is used to describe antibodies that are subject to the invention. However, antibodies may still possess some affinity for binding to cell surface antigens.
The ability of the non-cell binding antibody to induce an immunological response in a patient who is intended to be treated by the therapeutic antibody depends on the presence of at least one of the epitopes present in the therapeutic anti-body.
The noncell-binding antibodies are able to tolerate anti-idiotypic reactions, preferably at least the hypervariable V domain regions of the therapeutic antibodies and preferably to the framework region. It is therefore desirable that the tolerising antibodies have an amino acid pattern similar to that of the therapeutic antibody. It is preferred that the amino acid sequences between the variable domains in the non-cell binding antibody and the therapeutic antibodies are >90% identical, >95% identical or >99% identical. The differences should be limited to amino acid substitutions that are necessary to reduce the antigen-binding affinity of the non-cell binding antibody.
Preferred also, the non-cell binding antibody is capable to induce tolerance to the constant region of the therapeutic antibodies.” It is therefore preferred that the constant regions of the non cell-binding antibodies are similar to the therapeutic antibody. For example, they should have >90%, >95%, or >99% amino-acid sequence identity. The constant domains are identical in the non-cell binding antibody and therapeutic antibody, and thus are matched allotypically.
The invention also provides fragments of the antibody described herein that have tolerance-inducing capabilities. These fragments can be monovalent or divalent, such as Fab, Fab? These fragments include monovalent and divalent fragments such as Fab, Fab? )2 fragments. Single chain antibodies are also included. According to the invention, these fragments have preferred characteristics that are described in this document with respect to non-cell binding antibodies. These non-cell-binding antibodies may be used with therapeutic antibody fragments or therapeutic antibody molecules.
The reduced binding affinity for the non-cell-binding antibody can be achieved by a number of different methods. In the preferred embodiment, a change in the CDRs containing one, two, or three amino acid substitutes reduces the binding affinity. Alternatively, amino acids can be substituted in other parts of an antibody molecule to reduce the binding affinity. Amino acid substitutions within the framework regions, for example, are known to have a significant effect on binding affinity (Reichmann and al 1988). A monovalent therapeutic antibody is another alternative. Monovalent antibodies are less binding-affinity than their bivalent counterparts. Monovalent forms can be Fab fragments or single-chain antibodies or other genetically engineered fragments of antibody that retain a single binding site. Monovalent variants are also possible by changing the cysteine residue that participates in interchain disulphide (H-H). Monovalent antibodies may have a lower binding affinity than their bivalent counterparts, which could be enough to induce tolerance. The monovalent antibody should be incapable of binding Fc-receptors or C1q complement component, or both. These properties can be introduced either by mutation or both (see, for example, Morgan et. al., WO94/29351 published on 22 December 1994, and Winter et. al. EP0 307-434 B1).
The noncell-binding antibody or fragments as described in the invention can be any of several types of antibodies, including genetically modified antibodies or antibodies fragments. The antibodies or fragments are also generally derived from a variety of sources. They may, for example, be chimeric. Human constant regions with rat variables domains, or humanised, or CDR grafted, or otherwise reshaped. No. No. 0 125 023 B1; Boss et al., U.S. Pat. No. 4,816,397; Boss et al., European Patent No. 0 120 694 B1; Neuberger, M. S. et al., WO 86/01533; Neuberger, M. S. et al., European Patent No. 0 194 276 1; Winter, U.S. Pat. No. 5 225 539; Winter, European Patent No. 0 239 400 B1; Queen et al., U.S. Pat. No. 5,585,089; Queen et al., European Patent No. 0 451 216 B1; Adair et al., WO 91/09967, published 11 Jul. 1991; Adair et al., European Patent No. Adair et.al., European Patent No. 0 519 596 A1. Newman, R. and al. Biotechnology 10: 1455-1460, (1992), concerning primatized antibodies, as well as Huston, et. al. U.S. Pat. No. 5,091,513; Huston et al, U.S. Pat. No. 5,132,405; Ladner et al., U.S. Pat. No. No. Campath-1H has two amino acid changes in the framework region. It is still considered humanised.
Click here to view the patent on Google Patents.
Leave a Reply