Invented by Robert D. Bremel, Jane Homan, ioGenetics LLC
Bioinformatics is an interdisciplinary field that combines biology, computer science, and statistics to analyze and interpret biological data. In the context of peptide binding, bioinformatic methods aim to predict the binding affinity between a peptide and its target protein or receptor. These methods utilize various computational techniques, including machine learning, molecular docking, and molecular dynamics simulations, to predict the binding strength and identify key binding residues.
One of the main drivers of the market growth is the increasing need for peptide-based therapeutics. Peptides have gained significant attention in drug development due to their high specificity, low toxicity, and potential for targeting various diseases, including cancer, autoimmune disorders, and infectious diseases. However, the success of peptide-based drugs heavily relies on their binding affinity to the target protein. Bioinformatic methods provide a cost-effective and time-efficient approach to screen and optimize peptide candidates for drug development, reducing the need for expensive and time-consuming experimental assays.
Another factor contributing to the market growth is the availability of large-scale biological datasets. Advances in high-throughput technologies, such as next-generation sequencing and proteomics, have generated vast amounts of data on protein-peptide interactions. Bioinformatic methods can leverage these datasets to train predictive models and improve the accuracy of peptide binding predictions. Additionally, the integration of multiple data sources, such as protein structures, sequence motifs, and physicochemical properties, further enhances the predictive power of bioinformatic tools.
The market for bioinformatic methods for determining peptide binding is also driven by the increasing adoption of personalized medicine. Personalized medicine aims to tailor medical treatments to individual patients based on their genetic makeup, lifestyle, and environmental factors. Peptide binding predictions can aid in identifying patient-specific peptide targets and designing personalized therapies. Bioinformatic methods enable the analysis of large-scale genomic and proteomic data to identify peptide targets associated with specific diseases or patient subgroups, facilitating the development of targeted therapies with higher efficacy and fewer side effects.
Despite the promising growth prospects, there are several challenges that need to be addressed in the market for bioinformatic methods for determining peptide binding. One of the challenges is the limited availability of high-quality experimental data for training and validating predictive models. The accuracy of bioinformatic tools heavily relies on the quality and diversity of the training data. Efforts are being made to curate and standardize peptide binding datasets to improve the performance of predictive models.
Another challenge is the complexity of peptide-protein interactions. Peptide binding is influenced by various factors, including electrostatic interactions, hydrogen bonding, hydrophobic interactions, and conformational changes. Developing accurate bioinformatic methods that can capture the complexity of these interactions remains a challenge. Ongoing research focuses on integrating multiple computational approaches and improving the representation of peptide-protein interactions in predictive models.
In conclusion, the market for bioinformatic methods for determining peptide binding is witnessing significant growth due to the increasing demand for peptide-based therapeutics, the availability of large-scale biological datasets, and the adoption of personalized medicine. The development of accurate and efficient bioinformatic tools is crucial for accelerating drug discovery, optimizing peptide-based therapies, and advancing our understanding of peptide-protein interactions. As research and technological advancements continue, the market for bioinformatic methods for determining peptide binding is expected to expand further, offering new opportunities for innovation and growth in the field.

The ioGenetics LLC invention works as follows
This invention is a method for identifying peptide binding to ligands. It is particularly aimed at identifying epitopes expressed in mammalian and microorganism cells. The invention provides polypeptides containing the epitopes and vaccines and antibodies that use or are developed using these epitopes.

Background for Bioinformatic methods for determining peptide binding
Infectious disease, including those that were once thought to be controlled with antibiotics and vaccinations, continues to be a major cause of death worldwide. Currently, infectious and parasitic disease account for more than 15% of all deaths in the world. They are experiencing a resurgence due to increasing antimicrobial resistance and secondary complications of HIV AIDS. (World Health Organization Global Burden of Disease 2004, 2004). Climate change and an increase in population density will also likely lead to an increase in infectious diseases, as people are exposed to new environmental reservoirs. The pandemic H1N1 flu of 2009 is a good example of how a virus that can spread quickly and cause disease worldwide in a matter of months. “The threat of a genetically modified organism with equal transmissibility should also be a serious concern.
Antimicrobial Resistance is a growing problem worldwide. Certain antibiotic-resistant bacteria species are responsible for a disproportionate amount of morbidity, death and treatment costs. MRSA is the leading cause of nosocomial infection. Broad spectrum antibiotics are a major factor in the development of antimicrobial resistant bacteria. They place commensal and pathogenic flora under selective pressure. Broad spectrum antibiotics currently target a small number of metabolic pathways in bacteria. The majority of recently approved antimicrobials rely on the same pathways. This increases the risk of bioterrorism and the rapid development resistance. Clin. Infect. Dis. 38:1279-1286). “New strategies for antimicrobial research are urgently required that move beyond dependence on similar pathways and enable the elimination of specific pathogens while not placing selective pressure on antimicrobial flora in general.
In order to control infectious diseases using antibodies or vaccinations, it is necessary to characterize antigens and epitopes. There are several approaches to the characterization of epitopes. Immunologists began by producing monoclonal antibody or identifying antibodies from a patient’s serum bank, and then cloned epitopes using those antibodies. It is important to focus on immunodominant epitopes, while ignoring less dominant but more conserved epitopes. It has often led to the characterization of polysaccharide-based epitopes that are more susceptible to changing with growth conditions than genes-coded proteins. It is possible to obtain one or two epitopes that are characterized, which could offer protection against infection but may also be the ones most likely induce select pressure. This approach is purely antibody-based. Burnie et al. describe one such epitope characterization example. al. (Burnie et al. 2000. Infect. Immun. 68:3200-3209).
Reverse vaccinology is a field that starts with the genome, identifies open reading frames (ORFs) and proteins as vaccine components. Then they test their B-cell immunegenicity” (Musser J. M. 2006). Nat. Biotechnol. 24:157-158; Serruto, D., L. et al. 2009. Vaccine 27:3245-3250). Reverse vaccinology can be a powerful method, allowing rapid identification of proteins and potential epitopes from organisms that have a genome, regardless of whether the organism is easily cultured or not. The first reverse-engineered vaccine was developed against Neisseria Meningitidis by Pizza et. 2000. Science 287:1816-1820) is currently in Phase 3 trials, and similar efforts have been made on a variety of bacteria (Ariel and others). 2002. Infect. Immun. 70:6817-6827; Betts, J. C. 2002. IUBMB. Life 53:239-242; Chakravarti et al. 2000. Vaccine 19:601-612; Montigiani et al. 2002. Infect. Immun. 70:368-379; Ross et al. 2001. Vaccine 19:4135-4142.; Wizemann et al. 2001. Infect. Immun. 69:1593-1598). Pizza et. al., when identifying antigenic N. meningitides proteins in the proteome expressed concern that a small proportion of these antigenic protein could be expressed by E. coli due to their hydrophobicity because of transmembrane-domains. Rodriguez-Ortega was working with Strep. In order to isolate specific peptides, Rodriguez-Ortega has used the’shaving’ method with Strep. The surface loops of proteins can be removed with proteases in order to isolate peptides. 2006. Nat. Biotechnol. 24:191-197). This approach only harvests peptide loops that have at least two sites of protease cuts in the loop. As a result, it is unable to detect approximately 75% of surface peptides epitopes.
Diversity is a characteristic of all microorganisms. In nature, most species of bacteria are represented by many strains that look similar but may not be identical. Some of these strains have developed or lost certain metabolic traits like growth characteristics or antibiotic resistance. Some isolates differ antigenically and don’t offer cross protection against a subsequent infection. The level of variation between strains can vary from organism to organism. The most variable viruses are RNAs (including but not limited to influenza virus, foot and mouth, rotavirus, etc.) that undergo constant mutations and show constant antigenic drift, making vaccine selection difficult. “Identifying MHC high-affinity binding peptides, and B cell epitopes that are conserved across multiple strains is a challenge to epitope map.
Vaccines are not only for infectious diseases. Cancer vaccine therapies are currently being developed in Europe and America. These involve activating cytotoxic lymphocytes within the body of cancer patients by administering a tumor antigen. Clinical studies have reported positive results for specific tumor antigens. Subcutaneously administering melanoma gp100 antigen and intravascularly administering interleukin-2, for example, led to the reduction of tumors in 42% of melanoma patient. When the diversity of cancers are taken into consideration, it’s impossible to treat every cancer using a cancer vaccination consisting of a single type of tumor-antigen. The variety of cancer cells leads to a diversity in either the type of tumor antigens expressed or their amount. To develop new therapies, these antigens need to be identified. “We need new, more efficient ways to identify epitopes that can be used in the development of vaccines, diagnostics and therapeutics.
Autoimmunity is an immune response directed against the body’s cells. Autoimmunity may occur in several situations, including but not limited a failure to develop tolerance, exposure to an epitope that is normally shielded by the immune surveillance or as a result of exposure to exogenous antigens with characteristics similar to or mimicking the host cell’s MHC or binding characteristics. As a result of exposure to epitopes found in infectious agents that have mimics within the host tissue, autoimmune diseases are on the rise. “Rheumatic disease is a common sequela to streptococcal infections, diabetes type 1, and Guillain-Barre syndrome are all linked to Coxsackie or Rotavirus exposure.
Although the epitope structure is important for developing biotherapeutics and vaccines, it’s not the only thing that matters. It’s also necessary to understand the protein interactions involved in binding reactions. This includes, but is not limited to, enzymatic reaction, the binding of ligands with cell receptors, and other physiological mechanisms.
A mathematical approach that can be implemented in-silico to understand the structurally based peptide bonding mechanisms involved in immunologic reactions and other protein based reaction would be a great benefit to the art.
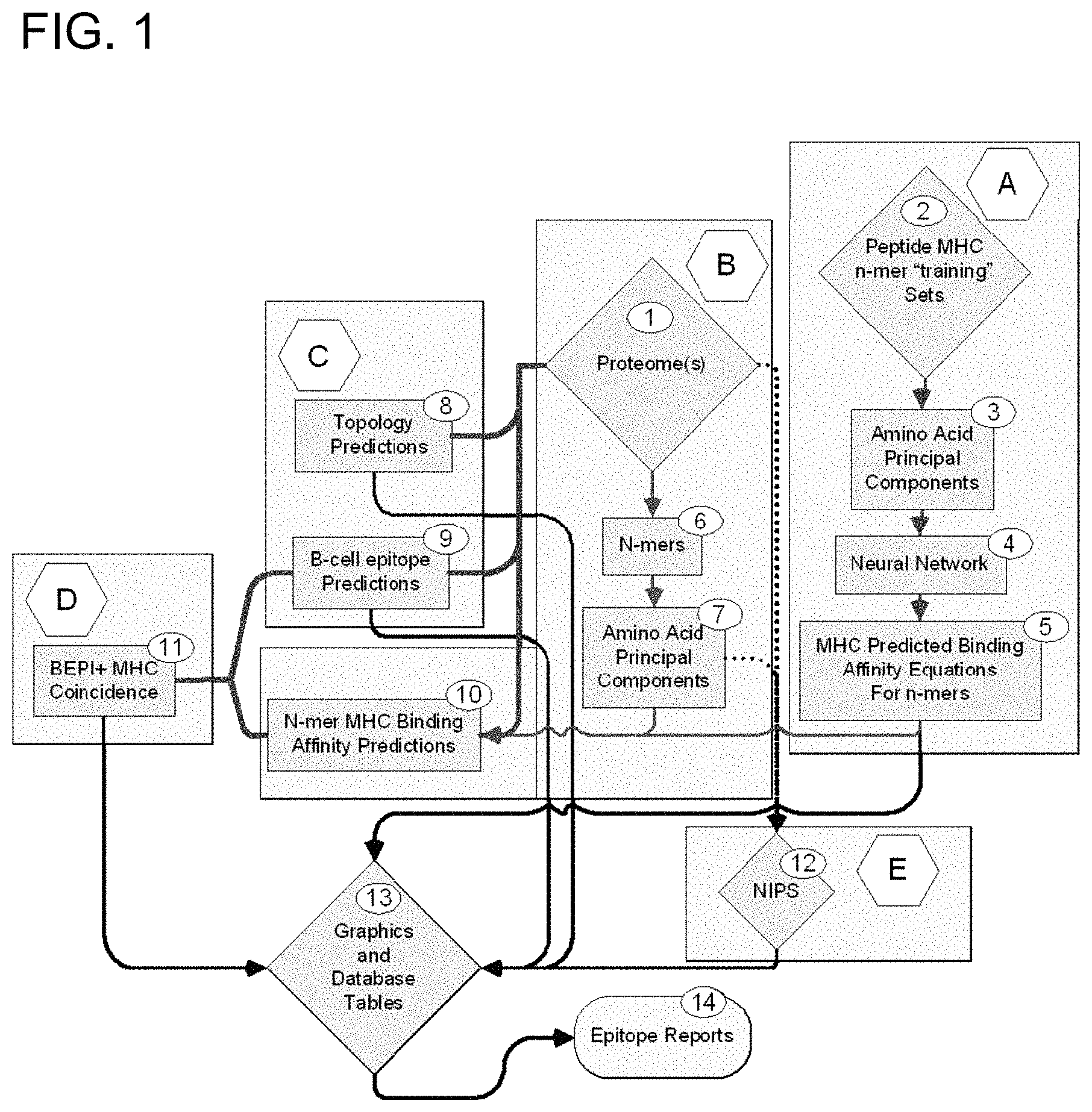
The present invention relates to a method of identifying in silico peptides or sets of peptides on the surface and/or internal to microorganisms that have a high likelihood of stimulating humoral immune responses and cell-mediated immunity. The method uses multiple predictive tools in order to produce a composite that includes both the topology of peptides and their binding or affinity properties. It is possible to characterize and predict specific peptides that are B-cell epitope sequences or MHC binding region in terms of their spatial and topological relationship. The present invention also identifies peptide sequences that have a high likelihood of being B cell and/or MHC sites containing T-cells epitopes, either on the surface or internal of a variety microorganisms, or cells. The binding sites are externally or internally located on the virion, or expressed on the virus-infected cell in some embodiments.

In some embodiments the present invention provides processes for identifying or analysing ligands. These processes are preferably computer-implemented and comprise: entering an amino acid sequence of a target into a processor; and analyzing multiple physical parameters of subsets amino acids of the sequence using a processor to identify subsets amino acids that interact with (e.g. bind) a binding partner (e.g. a B-cell receptor, antibody, or MHC I or MHC II binding region). In some embodiments the processes also include a mathematical formula to describe the subsets of amino acids. In some embodiments the mathematical expression is used to predict whether the amino acid subsets will bind with a binding partner. In some embodiments the processes also include generating sequences of amino acid subsets that have been identified as having affinity for a partner.
In some embodiments the binding partner may be an MHC-binding region. In certain embodiments, the binding partners is a B cell receptor or antibody. In certain embodiments, a peptide is used as a ligand that binds a MHC-binding region. In some embodiments the MHC-I region is used as a binding region. In some embodiments the MHC binding regions is a MHC II binding region. In certain embodiments, the polypeptide binds both to an MHC-binding region and a B cell receptor or antibody. In some embodiments the ligand is polypeptide which binds to B-cell receptors or antibodies. In some embodiments the length of an amino acid subset can range from 4 to 50 amino acids, or 4 to 30 amino acids, or 4 to 20 amino acids, or 5 to 15 amino aminos. In some embodiments the subsets are based on the n-terminus, where n is the initial amino acid and c the last amino. The sets consist of peptides ranging from 4 to 50 amino acids (or other ranges outlined above), starting at n, and continuing until n+1 ends up at c, for the length of peptides chosen. In some embodiments the amino acids are contiguous.
In some embodiments, the process further comprises constructing a network of neural connections via the computer. The neural networks are then used to predict binding affinity for one or more MHC-binding regions. In some embodiments the neural network can provide a quantitative relationship between structure and activity. In some embodiments the first three components of an amino-acid account for more than 80%.
In some embodiments the processes also include constructing a multilayer perceptron neuro network regression process, wherein the output for a peptide binding to an MHC binding area is LN(Kd). In some embodiments the regression process generates a series equations which allow the prediction of binding affinity based on the physical properties within the subsets. In some embodiments the regression process creates a series equations which allow binding affinity to be predicted using the physical properties within subsets of amino acids. In some embodiments the neural network’s performance when used with test peptides is statistically no different from random peptides at a 5% level. In some embodiments the processes also include utilizing a multi-layer perceptron with a number hidden nodes that correlates with the number of amino acid accommodated by an MHC binding area. In some embodiments the number of nodes hidden is between 8 and 60.
In some embodiments, a neural network is validated using a set of training affinities for peptides with known amino acid sequences. In some embodiments the neural network can be trained to predict binding of more than one MHC-binding region. In some embodiments the neural network generates a set equations that describe the physical properties and predict their contribution to Ln(Ki) for each subset. In some embodiments the equations are used to analyze peptide subsets that represent at least 25 percent of the target source’s proteome in order to determine the LN(kd). In some embodiments a standardization procedure is performed on raw data sets of binding affinity so that the characteristics of MHC molecules with different distributional properties can be directly compared. Standardization is the subtraction of the mean from all values in a set, and then the division of the result by the standard deviation. This produces a transformed variable that has a mean of 0 and a unit variance. These transformed data sets have a number desirable properties that are useful for statistical analysis.
In some embodiments, processes include the step of determining cellular locations of subsets of proteins, wherein cellular locations are selected from intracellular, extracellular or within a cell membrane. Signal peptides and combinations of these can also be used. In some embodiments extracellular peptides may be selected for testing and/or further analysis.
In some embodiments, the process further comprises the step of analyzing subsets polypeptides to predict B-cell epitopes. In some embodiments the processes also include constructing a network of neural cells via a computer. The neural network can then be used to predict the B-cell sequences. In some embodiments the processes include the step of comparing the properties of B-cell epitope and MHC. In certain embodiments, peptides with predicted B-cell sequence properties and MHC properties are selected for testing and/or further analysis. Extracellular peptides with predicted B-cell sequence properties and MHC properties may be selected for further testing and/or analysis in some embodiments. Secreted peptides with predicted B-cell sequence properties and MHC properties are selected in some embodiments for further testing and/or analysis. Extracellular peptides that are conserved between organism strains with predicted B-cell sequence properties or MHC binding property can be selected for further testing and analysis. In some embodiments, MHC-binding properties include a predicted affinity to at least one MHC-binding region chosen from the group consisting about greater 105 M1?1, greater 106 M1?1, greater 107 M1?1, greater than 810 M1?1 and greater than 1010 M1?1. In certain embodiments, processes include selecting peptides with binding affinity to at least one MHC binding region for further testing and/or analysis. In some embodiments the process includes selecting peptides with binding affinity for at least 2, 4, 10 20, 30, 40, 60, 70 or 80 MHC regions, or 1 to 5, 1-10, 1-20, 5-10, 5-10, 5-10, 10-20, 10-20, 10-30 or 10-50, to be used in further analysis or testing. In some embodiments the processes also include selecting peptides with defined MHC properties. The MHC properties are defined as having a predicted affinities for at least 1, 2, 3, 4, 10, 20, 30, 60, 70 or 80 or 100 or more MHC regions or 1 to 5, 1-10, 1-20, 5-10, 5-20, 10-20, 10 to 20 or 10-20, 10-30 or 10-50 MHC-binding regions.
In some embodiments the physical properties can be used to predict the binding affinity of a B cell receptor or antibody. In certain embodiments, the process further comprises constructing a network of neural connections via the computer. The neural network can then be used to predict binding affinity for one or more antibodies or B-cell receptors. In some embodiments the process further comprises the step of selecting the peptides that have a binding affinity for the B-cell receptors and antibodies. In certain embodiments, physical properties can be used to predict the binding affinity of a receptor. In some embodiments the process further comprises constructing a network of neural connections via the computer. The neural networks are then used to predict binding affinity to a receptor. In some embodiments the processes include the step of selecting the peptides that have a high affinity for the receptor cellular.
In some embodiments, the amino acid sequence comprises the amino acid sequences of a class of proteins selected from the group consisting of membrane associated proteins in the proteome of a target source, secreted proteins in the proteome of a target organism, intracellular proteins in the proteome of a target source, and viral structural and non-structural proteins. In some embodiments, the process is performed on at least two different strains of a target organism. In some embodiments, the target source is selected from the group consisting of prokaryotic and eukaryotic organisms. In some embodiments, the target source is selected from the group consisting of bacteria, archaea, protozoas, viruses, fungi, helminthes, nematodes, and mammalian cells. In some embodiments, the mammalian cells are selected from the group consisting of neoplastic cells, carcinomas, tumor cells, cancer cells, and cells bearing an epitope which elicits an autoimmune reaction. In some embodiments, the target source is selected from the group consisting of an allergen, an arthropod, a venom and a toxin. In some embodiments, the target source is selected from the group consisting of Staphylococcus aureus, Staphylococcus epidermidis, Cryptosporidium parvum and Cryptosporidium hominis, Mycobacterium tuberculosis, Mycobacterium avium, Mycobacterium ulcerans, Mycobacterium abcessus, Mycobacterium leprae, Giardia intestinalis, Entamoeba histolytica, Plasmodium spp, influenza A virus, HTLV-1, Vaccinia and Rotavirus. In some embodiments, the target source is an organism identified in Tables 14A or 14B.
In some embodiments at least 80% possible subsets of amino acids within an amino acid sequence with a length of n are analysed, where n ranges from approximately 4 to 60. In some embodiments the amino acid subset can be conserved between multiple strains. In some embodiments multiple strains can be selected from a group of 3, 5, 10, 20, 30, 40, 60, or 100 strains.

Click here to view the patent on Google Patents.
Leave a Reply